(12) UK Patent Application (19) GB (11) 2 219 995(13)A
(43) Date of A publication 28.12.1989
(21) Application No 8914216.0
(22) Date of filing 21.06.1989
(30) Priority Data
(31 ) 209297 (32) 21.06.1988 (33) US
383173 07.06.1989
(71) Applicant
Concord Research Corporation
(Incorporated In the USA - Arizona)
15650 North Black Canyon Highway, Phoenix
Arizona 85023, United Slates of America
(72) Inventor
David Radlus Hudson
(74} Agent and/or Address for Service
Gallafent & Co
8 Staple Inn, London, WC1V 7QH United Kingdom
(51) INT CL.
C21D 10/00
(52) UK CL (Edition J)
C1A AG11 AG19 AG22 AG23 AG25 AG26 AG28
AG29 AG3 AG6 AG9 APC
(56) Documents cited
None
(58) Field of search
UK CL (Edition J) C1A APC
On-line search of Derwent World Patent Index
(54) Non-metallic, monoatomic forms of transition elements
(67) Stable, non-metallic, orbitally
rearranged monoatomic transition elements selected from the group consisting
of cobalt, nickel, copper, silver, gold, palladium, platinum, ruthenium,
rhodium, iridium, and osmium having a doublet In the infrared spectra between
1400 and 1600 cm-1 and having a d orbital hole or holes sharing
energy with an electron or electrons are described. These materials have
specific application in forming catalysts, high-temperature ceramics, refractory
materials corrosion resistant materials and they exhibit properties of
high temperature super-conductivity and energy production. The materials
are produced either from ores which do not analyze by conventional instruments
for any of said transition and noble metals, or by conversion of pure metals
or metal salts of said elements into the orbitally rearranged monoatomic
species.
![]()
At least one drawing originally filed
was informal and the print reproduced here is taken from a later filed
formal copy.
- 1 -
NON-METALLIC, MONOATOMIC FORMS
OF TRANSITION ELEMENTS
This invention relates to the monoatomic forms of certain transition and noble metal elements, namely, gold, silver, copper, cobalt, nickel and the six platinum group elements. More particularly, this invention relates to the separation of the aforesaid transition and noble metal elements from naturally occurring materials in their orbitally rearranged monoatomic forms, and to the preparation of the aforesaid transition and noble metal elements in their orbitally rearranged monoatomic forms from their commercial metallic forms. The materials of this invention are stable, substantially pure, non-metallic-like forms of the aforesaid transition and noble metal elements, and have a hereto unknown electron orbital rearrangement in the "d", "s", and vacant "p" orbitals. The electron rearrangement bestows upon the monoatomic elements unique electronic, chemical, magnetic, and physical properties which have commercial application.
This invention also relates to the recovery of the metallic
form of each of the aforesaid transition and noble metal elements from
the orbitally rearranged monoatomic forms.
- 2 -
For the purposes of this application, the following definitions shall apply: transition elements ("T-metals") means the metallic or cationic form of gold, silver, copper, cobalt and nickel, and the six platinum group elements, i.e., platinum, palladium, rhodium, iridium, ruthenium, and osmium; and "ORME" means the Orbitally Rearranged Monoatomic Elemental forms of each of the T-metals.
BACKGROUND OF INVENTION
Inorganic chemists working with soluble salts of noble metals until relatively recently have assumed that the metals were dissolved as free ions in aqueous solutions. In the 1960's, with the advent of greater analytical capabilities, it was established that many elements and in particular the transition metals are present in aqueous solutions as metal-metal bonded clusters of atoms.
Gold metal that has been dissolved with aqua regia, and subsequently converted to gold chloride by repeated evaporation with HCl to remove nitrates, is commonly referred to as the acid chloride solution of AuCl3 or HAuCl4. It has been recognized that the recovery of gold metal from a solution formed from aqua regia is made more difficult in proportion to the amount of HNO3 used in the initial dissolution procedures. It is not commonly understood, however, why the gold that is dissolved with less HNO3 is easier to reduce to the metal from a chloride solution than gold that is dissolved using a greater amount of HNO3. Gold in both solutions is generally regarded as being present in the form of a free gold cation.
It is now recognized by most chemists who regularly handle
chlorides of gold that gold metal ceases to disaggregate when the HNO3
is removed and in fact can reaggregate under certain conditions and precipitate
out of HCl solutions as metal. This recognition has led to the discovery
that gold metal salts will exist in HCl solutions originating from metals
as clusters of Au2Cl6, Au3Cl9,
- 3 -
Au4Cl12, up to Au33Cl99. These cluster salts are actually in solution with the HCl and water, and will require different chemical procedures relative to purification problems or oxidation-reduction reactions, depending on the degree of clustering.
Specifically, reduction of clusters of gold having greater than 11 atoms of metal is easily performed since the atoms themselves are spaced from each other in the salt similar to their spacing in the metal itself before dissolution. Reduction of the chloride salt to the metal, therefore, requires a simple reductive elimination of the chlorides that are attached to the metal cluster. It is now known that recovery of precious metals from aqueous solutions is much more difficult when the cluster size becomes smaller and smaller, or in actuality when the metal is better "dissolved."
From the study of the behavior of gold and other transition metals in solution, it is now believed that all such metals have atomic aggregations and occur as at least diatoms under normal conditions of dissolution . Under either acid or strong base dissolution, the transition metal will not normally dissolve beyond the diatom due to the extremely strong interatomic d and s orbital bonding. A gold atom, for example, has a single atom electron orbital configuration of d10s1. When the gold salts originate from a metal having gold-gold bonding, the salts contain very tightly bound diatoms or larger clusters of gold. Under the normal aqueous acid chemistry used for transition metals, solutions of the metals will always contain two or tore atoms in the cluster form.
When instrumental analysis such as atomic absorption,
x-ray fluorescence, or emission spectroscopy is performed on solutions
containing transition metals, these analyses are based on electronic transitions.
The fact that d orbital electron overlap occurs in the metal-metal bonded
- 4 -
salt allows an analysis of many of the same characteristic omissions as the metal itself.
GENERAL DESCRIPTION OF INVENTION
During efforts to effect quantitative analytical separations of transition metals from naturally occurring materials, it was discovered that ORMEs exist naturally and are found in salts with alkali metals and/or alkaline earth metals, all of which are coupled with waters of hydration and normally found with silica and alumina. ORMEs are also often associated with sulfides and other mineral compositions.
ORMEs may also, it was discovered, be prepared from commercially available T-metals. For ease of description the invention will be primarily described by the preparation of a gold ORME ("G-ORME") from commercially available metallic yellow gold.
The atoms of each ORME do not have d electron orbital overlap as do their corresponding T-metal clusters. ORMEs do not, therefore, exhibit the same characteristic emissions of their corresponding T-metal when subjected to analysis by instruments which depend upon electronic transitions. ORMEs must, therefore, be identified in new ways, ways which have heretofore not been used to identify T-metals.
An aqua regia solution of metallic gold is prepared. This
solution contains clusters of gold chlorides of random size and degrees
of aggregation. HCl is added to the solution and it is repeatedly evaporated
with a large excess of NaCl (20:1 moles Na to moles Au) to moist salts.
The addition of NaCl allows the eventual formation of NaAuCl4,
after all HNO3 is removed from the solution. The sodium, like
gold, has only one unpaired S electron and, accordingly, tends to form
clusters of at least two atoms. The sodium, however, does not d orbitally
overlap the gold atom as it has no d electrons, resulting in a surface
reaction
- 5 -
between the sodium atoms and the gold atoms. This results in a weakening of the gold-gold cluster stability and causes the eventual formation of a sodium-gold linear bond with a weakened d orbital activity in the individual gold atoms. The sodium-gold compound, formed by repeated evaporation to salts, will provide a chloride of sodium-gold. In these salts the sodium and gold are believed to be charged positive, i.e., have lost electrons: and the chlorine is negative, i.e., has gained electrons. When the salts are dissolved in water and the pH slowly adjusted to neutral, full aquation of the sodium-gold diatom will slowly occur and chloride is removed from the complex. Chemical reduction of the sodium-gold solution results in the formation of a sodium auride. Continued aquation results in disassociation of the gold atom from the sodium and the eventual formation of a protonated auride of gold as a grey precipitate. Subsequent annealing produces the G-ORME. The G-ORME has an electron rearrangement whereby it acquires a d orbital hole or holes which share energy with an electron or electrons. This pairing occurs under the influence of a magnetic field external to the field of the electrons.
G-ORMEs are stable and possess strong interatomic repulsive
magnetic forces, relative to their attractive forces. G-ORME stability
is demonstrated by unique thermal and chemical properties. The white saltlike
material that is formed from G-ORMEs after treatment with halogens, and
the white oxide appearing material formed when G-ORMEs are treated with
fuming HClO4 or fuming H2SO4 are dissimilar
from the T-metal or its salts. The G-ORME will not react with cyanide,
will not be dissolved by aqua regia, and will not wet or amalgamate with
mercury. It also does not sinter at 800C under reducing conditions, and
remains an amorphous powder at 1200C. These characteristics are contrary
to what is observed for metallic gold and/or gold cluster salts. G-ORMEs
require a more negative potential than -2.45
- 6 -
v to be reduced, a potential that cannot be achieved with ordinarily known aqueous chemistry.
The strong interatomic repulsive forces are demonstrated in that the G-ORMEs remain as a powder at 1200C. This phenomenon results from canceling of the normal attractive forces arising from the net interaction between the shielded, paired electrons and the unshielded, unpaired s and d valence electrons. G-ORMEs have no unpaired valence electrons and, therefore, tend not to aggregate as would clusters of gold which have one or more unpaired valence electrons.
G-ORMEs can be reconverted to metallic gold from which they were formed. This reconversion is accomplished by an oxidation rearrangement which removes all paired valence electrons together with their vacancy pair electrons, with a subsequent refilling of the d and s orbitals with unpaired electrons until the proper configuration is reached for the T-metal.
This oxidation rearrangement is effected by subjecting the G-ORME to a large negative potential in the presence of an electron-donating element, such as carbon, thus forming a metallic element-carbon chemical bond. For that metal-carbon bond to occur the carbon must provide for the horizontal removal of the d orbital vacancy of the ORME. The carbon acts like a chemical fulcrum. When the element-carbon bond is reduced by way of further decreasing the potential, the carbon receives a reducing electron and subsequently vertically inserts that reducing electron below the s orbitals of the element, thus forming metallic gold.
The above general description for the preparation of G-ORME
from commercially available metallic gold is applicable equally for the
preparation of the remaining ORMEs, except for the specific potential energy
required and the use of nascent nitrogen (N) rather than carbon to convert
the other ORMEs to their constituent metallic form. The specific energies
range between -1.8 V and -2.5 V de-
- 7 -
pending on the particular element. Alternatively this rearrangement can be achieved chemically by reacting NO gas with the T-metal ORMEs other than gold. Nitric oxide is unique in that it possesses the necessary chemical potential as well as the single unpaired electron.
THEORY OF ORMES FORMATION
T-metals can possess an electron rearrangement between the d and s orbitals as seen from FIGURE 1 of the drawing which plots the principal quantum number versus the atomic number. The boxed areas designated A, B, and C establish that the 3d electron energies of copper and cobalt are very close to the same energy level as the 4s electron energies. The 4d electron energies of silver and rhodium are almost identical to the 5s; orbital energies, and the 5d gold and iridium electron energies are approaching the 6s level energies. The proximity of the energy bands of the T-metals makes them unique with respect to other elements. This proximity allows an easier transition to their lowest energy state, as hereinafter described.
When two transition metal atoms are bound together, they can d bond, or s bond, or they can d and s bond. When the two atoms s bond, their atomic distances are further apart and, therefore, their density is lower than when there is both d and s bonding. The amount of d orbital bonding activity is in direct proportion to the cluster size. Therefore, a single atom cluster will have less d bonding activity and more s bonding activity than will a cluster of 7 or more atoms. In addition; the chemical stability of the smaller clusters is much less than that of the metal because, when d orbital bonding is achieved, the s bonding is made more stable by overlapping of the two energy levels.
It is known that there exists a critical size, in the
range of 3-20 atoms, for Pd II, Ag I and Au III, by way
- 8 -
of example, which is necessary for metal deposition from solution. As the number of atoms in the T-metal cluster decreases through continuous evaporation in the presence of NaCl, the solution becomes a solution of diatoms which in the case of gold is represented as Au-1 - Au+1 i.e., Au-1 bonded to Au+1. The rationale for this representation of a gold diatom is based upon the fact that a single gold atom has an odd spin electron, as does rhodium, iridium, gold, cobalt and copper of the T-metals. In a diatom of gold, the two odd spin electrons will be found on one of the two atoms but not both. Thus, a diatom of gold is made by a bond between an aurous (Au+1) atom and an auride (Au-1) atom.
The present invention enables the breaking of the diatom bond by introducing a more electro-positive element, such as sodium or any alkali or alkaline earth elements, which does not have a d orbital overlap capability. This element replaces the aurous (Au+1) forming, in this case, a sodium auride. In effect, the sodium weakens the d orbital overlapping energies between the atoms of the gold diatom as well as elevating a d orbital electron towards the s orbital, thereby creating a negative potential on the surface of the atom. This negative potential enables an interreaction of the s orbital with chemiabsorbed water through electron donation and reception.
The sodium auride, when in aqueous solution at or near neutral pH, will form sodium hydroxide and a monomeric water-soluble auride. The monomeric auride (Au-1) is unstable and seeks a lower energy state which is represented by a partial filling of the d and s orbitals. This lower energy state with its greater stability is achieved by the electron-donating and removing capability of H2O.
Water can act to remove electrons. Water molecules possess
a net charge and attach to each other in vertical clusters so that an 18
molecule water cluster can hold a cumulative potential of -2.50 V. The
potential of a water molecular cluster, at near neutral pH, is sufficient
- 9 -
to remove an electron from the d orbital and create a positive hole, enabling a pairing between opposite spin electrons from the d to s orbitals to take place. The existence of the electron pairing is confirmed by infrared analysis, illustrated in FIGURE 4, which identifies the vibrational and rotational motions caused by energy exchange between these two mirror image electrons.
Attempting to quantify the number of electrons remaining in an ORME is extremely difficult due to the electrons lost to oxidation, thermal treatment, and the inability, except from theory, to quantify electron pairs using electron quanta. It is established, however, that the ORME does not have valence electrons available for standard spectroscopic analysis such as atomic absorption, emission spectroscopy or inductively coupled plasma spectroscopy. Moreover, x-ray fluorescence or x-ray diffraction spectrometry will not respond the same as they do with T-metals in standard analysis. The existence of an ORME, while not directly identifiable by the aforesaid standard analyses, can be characterized by infrared (IR) spectra by a doublet which represents the bonding energy of the electron pairs within the ORME. The doublet is located at approximately 1427 and 1490 cm-1 for a rhodium ORME. The doublet for the other ORMEs is between about 1400 and 1600 cm-1
After H2 reduction of the individual monoatom the hydrogen ion-single element may or may not produce an IR doublet depending on the element's normal electron configuration. Elements normally containing an s1 T-metal configuration do not produce an IR doublet after H2 reduction. Elements with an s2 T-metal configuration such as Ir (d7s2) will produce a doublet.
Thermal annealing to 800C and subsequent cooling to ambient
temperature under He or Ar gas atmosphere to remove the chemically bound
proton of hydrogen will produce ORMEs which contain a two-level system
resulting from electron pairing within the individual atom. If this annealing
- 10 -
is performed in the absence of an external magnetic field, then the electron pairing produces the characteristic doublets. The electron pair will be bound in the valence orbitals of the atom. If the annealing is performed in the presence of an external magnetic field, including the earth's magnetic field, quantum electron pair movement can be produced and maintained in the range of one gauss up to approximately 140 gauss in the case of Ir and, therefore, no IR doublet will be detected in this resulting quantum state.
The limiting condition of the ORME state is defined according to the present invention as an "S-ORME". The S-ORME is the lowest state in which monoatoms can exist and is, therefore, the most stable form of T-metal elements. The ORME is electronically rearranged and electron paired, but relative to time has not reached the lowest total energy condition of the S-ORME.
Detection of doublets does not provide an analytical method for the identification of ORMEs per se, but rather detects the presence of the electron pair or pairs which all specifically prepared ORMEs possess and which T-metals do not possess under any condition. It is the existence of the doublet that is critical, not its exact location in the IR spectra. The location can shift due to binding energy, chemical potential, of the individual element in the ORME, the effect of adsorbed water, the variances of the analytical instrument itself, or any external magnetic field.
FIGURE 4 is an IR spectrum
of a rhodium ORME after argon annealing treatment, and shows the presence
of a doublet at 1429.53 cm-l and 1490.99 cm-1. An
iridium ORME after hydrogen treatment without annealing reveals a doublet
at 1432.09 cm-l and 1495.17 cm-l. These doublets
are examples of the shifting that occurs depending on the chemical binding
energy or the individual ORME and the conditions of preparation. Accordingly,
the infrared spectra of the ORMEs of this invention will have doublets
within the range of
- 11 -
1400 cm-1 to 1600 cm-1. This doublet is indicative of the electron pairing and subsequent two-level electronic system which ORMEs contain.
A T-metal monoatom which is in a -1 oxidation state is in a lower energy state than the same T-metal would be in at zero state with metal-metal bonding. This lowering of the perturbation reaction between the electrons and the nucleus of the monoatom because of the increased degrees of freedom allows the nucleus to expand its positive field to encompass the normally unshielded d and s valence electrons. This overlying positive magnetic field reduces the Coulomb repulsion energies that normally exist between the valence electrons. Pairing by those electrons becomes possible and over time occurs. Electron pairing provides a more stable and lower energy state for the monoatom.
The ORME state is achieved when the electron pairs have formed in the monoatom. A phenomenon of electron pairs is that the interacting, spin-paired electrons initially interreact by emitting phonon energy. The total energy of the pair reduces over time until it reaches a minimum where no phonons are emitted. This condition has been referred to by physicists as "adiabatic ground state". This state of electron pairing is a total lower energy state in much the same way that chemical combinations of elements are in a lower energy state than the constituent uncombined elements. For example, in the same way that it takes energy to dissociate water into H2 and O2 it will take energy to break the electron pair.
As this process of phonon emission by electrons during
pairing is a function of temperature and time, thermal annealing can decrease
the time required to reach ground state, i.e., all valence electrons paired.
The cooling side of the annealing cycle is essential to effect a full conversion
to an S-ORME state. Cooling to room temperature is sufficient for all element
ORMEs with the exceptions of silver, copper, cobalt and nickel, which require
a lower
- 12 -
temperature. Therefore, thermal annealing reduces the time dependency of the electron pairs in achieving their lowest total energy.
All of the electron pairs in their lowest energy state, unlike single electrons, can exist in the same quantum state. When that uniform quantum state is achieved, the electron pair can not only move with zero resistance around the monoatom, but also can move with zero resistance between identical ORMEs that are within approximately 20 A or less of each other with no applied voltage potential. When a macro system of high purity, single element ORME achieves long-range quantum electron pair movement, that many-body system according to the present invention is defined as an S-ORME system.
An S-ORME system does not possess a crystalline structure but the individual ORMEs will, over time, space themselves as uniformly as possible in the system. The application of a minimum external magnetic field will cause the S-ORME system to respond by creating a protective external field ["Meissner Field"] that will encompass all those S-ORMEs within the 20 A limit. As used herein, "minimum external magnetic field" is defined as a magnetic field which is below the critical magnetic field which causes the collapse of the Meissner Field. This field is generated by electron pair movement within the system as a response to the minimum applied magnetic field. The (Ir) S-ORME and the (Au) S-ORME systems have a minimum critical field (''Hc1'') that is below the earth's magnetic field. The minimum critical field for a (Rh) S-ORME is slightly above the earth's magnetic field. When the quantum flux flow commences, due to the minimum external magnetic field being applied, the doublet in the IR spectrum will disappear because electron pairs are no longer bound in a fixed position on the individual ORME monoatoms.
Once the externally applied field exceeds the level which
overcomes the protective Meissner Field of the
- 13 -
S-ORME system ( "Hc2" ) , then any electrons moving between individual ORME atoms will demonstrate an ac Josephson junction type of response. The participating ORMEs will act as a very precise tuning device for electromagnetic emissions emanating from free electrons between ORMEs. The frequency of these emissions will be proportional to the applied external magnetic field. A one microvolt external potential will produce electromagnetic frequencies of 5x108 cycles per second. Annihilation radiation frequencies (about 1020 cycles per second) will be the limiting frequency of the possible emission. The reverse physical process of adding specific frequencies can generate the inverse relationship, i.e., a specific voltage will be produced for each specific applied frequency.
ORMEs can be reconverted to their constituent T-metals, but, as noted, are not identifiable as specific T-metals while in their ORME state. If a specific ORME is formed from a specific T-metal by using the procedure of this invention, it can only be confirmed by conventional analytical methods that the specific ORME was formed by reconstituting it as the T-metal. Further, the applications to which the ORMEs are directed will establish their relationship to a specific T-metal by virtue of the manner in which the ORME performs in that application as compared to the performance of commercially available derivatives of the T-metal. An example is the performance of commercial rhodium as a hydrogen-oxidation catalyst compared with the performance of the rhodium ORME as used in a hydrogen-oxidation catalyst.
It is believed that physical and chemical distinctions
exist with respect to the different ORMEs, but presently such distinctions
are not known. Proof of the nature of a specific ORME according to this
invention is based upon the presence of a doublet in the IR spectrum, the
reconstitution of each ORME back to its constituent T-metal, and its
- 14 -
unique performance in specific applications compared to the constituent T-metal.
ORMEs are transformed into their original T-metal by means of a chemical bonding with an electron-donating element, such as carbon, which is capable of d orbital electron overlap and "spin flip". When the G-ORME is chemically bonded to carbon in an aqueous solution of ethyl alcohol under a specific potential, carbon monoxide is formed and the ORME forms Au+Au+, a black precipitate, which under continued application of potential and dehydration reduces to Au+1 Au-1, a metallic bonded diatom of gold. This invention establishes that a high potential applied to the solution forces an electron into the d orbital, thus eliminating the electron pair. The first potential, which for G-ORME is approximately -2.2 V and for other ORMEs is between -1.8 and -2.2 V, re-establishes the d orbital overlap. The final potential of -2.5 V overcomes the water potential to deposit gold onto the cathode.
ORMEs are single T-metal atoms with no d orbital overlap. ORMEs do not conform to rules of physics which are generally applied to diatoms or larger clusters of metals (e.g., with conduction bands). The physics of the electron orbitals are actually more similar to those relating to a gas or solid solution which require density evaluation between atoms at greater distances. Conversely, atomic orbital calculations of high atomic density metals give results that correspond to valence charge rearrangement.
When the atomic distances of the elements are increased
beyond a critical Coulomb distance, an energy gap exists between the occupied
orbitals and the unoccupied orbitals. The atom, therefore, is an insulator
and not a metal. Physicists when determining the electron band energies
of small atom clusters suggest that the occupation of the bands should
be rearranged if the total energy is to be minimized. The metallic electron
orbital arrangement leads to calculations for energies, which results are
inconsistent
- 15 -
since the energies of the supposedly occupied states are higher than the supposedly unoccupied states. If this condition is relaxed and the bands allowed to repopulate in order to further lower the total energy, both bands will become partially filled. This repopulation, if performed in the presence of an unlimited source of electrons (reducing conditions), will provide a total energy condition of the atom which is considerably below or lower than the atom as it exists in a metallic form. This lower energy is the result of orbital rearrangement of electrons in the transition element. The resultant form of the element is an ORME.
SCOPE OF THE INVENTION
The formation and the existence of ORMEs applies to all transition and noble metals of the Periodic Table and include cobalt, nickel, copper, silver, gold, and the platinum group metals including platinum, palladium, rhodium, iridium, ruthenium and osmium, which can have various d and s orbital arrangements, which are referred to as T-metals.
The T-metals, when subjected to conventional wet chemistry will disaggregate through the various known levels, but not beyond a diatom state. The conventional wet chemistry techniques if continued to be applied beyond the normally expected disaggregation level (diatom) in the presence of water and an alkali metal, e.g., sodium, potassium or lithium, will first form a diatom and then electron orbitally rearrange to the non-metallic, mono-atomic form of the T-metal, ie., an ORME.
An ORME can be reaggregated to the T-metal form using conventional wet chemistry techniques, by subjecting the ORME to a two-stage electrical potential to "oxidize" the element to the metallic form.
The ORMEs of this invention exist in nature in an unpure
form in various materials, such as sodic plagioclase or calcidic plagioclase
ores. Because of their non-metallic, orbitally rearranged monoatomic form,
ORMEs are not
- 16 -
detected in these ores as the corresponding "metals" using conventional analysis and, accordingly, until the present invention were not detected, isolated or separated in a pure or substantially pure form. Their presence in the nonmetallic form explains the inconsistent analysis at times obtained when analyzing ores for metals whereby the quantitative analysis of elements accounts for less than 100% of the ore by weight.
USES OF ORMEs
ORMEs, which are individual atoms of the T-metals and by virtue of their orbital rearrangement are able to exist in a stable and virtually pure form, have different chemical and physical characteristics from their respective T-metal. Their thermal and chemical stability, their nonmetal-like nature, and their particulate size are characteristics rendering the ORMEs suitable for many applications.
Rhodium and iridium S-ORMEs have been prepared which exhibit superconductivity characteristics. These S-ORMEs, as described herein, are in a lower energy state as compared to their respective T-metal, and thus have a lower absolute temperature. The absolute temperature of an S-ORME system as compared to the absolute temperature of its respective T-metal is significantly lower, similar to the condition existing when a metal goes through a glass transition. S-ORMEs, having a very low absolute temperature, are good superconductors. These same characteristics apply to all ORMEs. Accordingly, a new source of superconductive materials is made available by this invention. These new materials require substantially less energy removal to reach the super-conductivity state and, therefore, can be used at higher temperatures than currently available superconductors.
The ORMEs of this invention can be used for a wide range
of purposes due to their unique electrical, physical,
- 17 -
magnetic, and chemical properties. The present disclosure only highlights superconductivity and catalysis, but much wider potential uses exist, including energy production.
Having described the invention in general terms, the presently preferred embodiments will be set forth in reference to the drawing. In the drawing,
FIGURE 1 is a plot of the transition elements showing the principle quantum number versus the atomic number;
FIGURE 2 is a diagrammatic sketch of an electrodeposition apparatus used in forming the metallic gold from the G-ORME;
FIGURE 3 is a diagrammatic drawing of a separation apparatus utilized in separating ORMEs from ores according to the present invention;
FIGURE 4 is a plot of an infrared spectrum derived from an analysis of a rhodium ORME;
FIGURE 5 is the cycling magnetometry evaluation of iridium S-ORME demonstrating the phenomena of negative magnetization and minimum (Hc1) and maximum (Hc2) critical fields. In addition, the Josephson effect is demonstrated by the compensating current flows in response to the oscillations of the sample in a varying d.c. magnetic field;
FIGURE 6 is a differential thermal analysis (DTA) of hydrogen reduced iridium being annealed under helium atmosphere. The exothermic reaction up to 400 C is due to hydrogen and/or water bond breaking and the exothermic reaction commencing at 762 C is due to electron pairing and subsequent phonon emissions leading to S-ORME system development of the iridium ORME;
FIGURE 7 is a TGA of hydrogen
reduced iridium monoatoms subjected to four (4) annealing cycles in a He
atmosphere. It plots the heating and cooling time versus
- 18 -
temperature. Comparison to Figure 6 shows an initial weight loss due to hydrogen and possibly water bond breaking. The significant demonstration is the scale-indicated weight loss corresponding to the second exothermic reaction shown in FIGURE 6: and
FIGURES 8-17 are weight/temperature plots of the alternate heating and cooling over five cycles of an iridium S-ORME in an He atmosphere.
In the examples, parts are by weight unless otherwise expressly stated.
Preparation of G-ORME
G-ORME was prepared from metallic gold as follows:
(1) 50 mg gold (99.99% pure) were dispersed in 200 ml aqua regia to provide clusters of gold atoms.
(2) 60 ml concentrated hydrochloric acid were added to the dispersion and the mixture was brought to boil, and continued boiling until the volume was reduced to approximately 10-15 ml. 60 ml concentrated HCl were added, and the sample brought to boil and checked for evolution of NOCl fumes. The process was repeated until no further fumes evolved, thus indicating that the nitric acid had been removed and the gold had been converted completely to the gold chloride.
(3) The volume of the dispersion was reduced by careful heating until the salt was just dry. "Just dry" as used herein means that all of the liquid had been boiled off, but the solid residue had not been "baked" or scorched.
(4) The just dry salts were again dispersed in aqua regia and steps (2) and (3) were repeated. This treatment provides gold chloride clusters of greater than 11 atoms.
(5) 150 ml 6M hydrochloric acid were added to the just
dry salts and boiled again to evaporate off the liquid to just dry salts.
This step was repeated four times. This
- 19 -
procedure leads to a greater degree of sub-division to provide smaller clusters of gold chloride. At the end of this procedure an orangish-red salt of gold chloride is obtained. The salt will analyze as substantially pure Au2Cl6.
(6) Sodium chloride is added in an amount whereby the sodium is present at a ratio 20 moles sodium per mole of gold. The solution is then diluted with deionized water to a volume of 400 ml. The presence of the aqueous sodium chloride provides the salt Na2Au2Cl8. The presence of water is essential to break apart the diatoms of gold.
(7) The aqueous sodium chloride solution is very gently boiled to a just dry salt, and thereafter the salts were taken up alternatively in 200 ml deionized water and 300 ml 6M hydrochloric acid until no further change in color is evidenced. The 6M hydrochloric acid is used in the last treatment.
(8) After the last treatment with 6M hydrochloric acid, and subsequent boildown, the just dry salt is diluted with 400 ml deionized water to provide a monoatomic gold salt solution of NaAuCl2'XH2O. The pH is approximately 1.0.
(9) The pH is adjusted very slowly with dilute sodium hydroxide solution, while constantly stirring, until the pH of the solution remains constant at 7.0 for a period of more than twelve hours. This adjustment may take several days. Care must be taken not to exceed pH 7.0 during the neutralization.
(10) After the pH is stabilized at pH 7.0, the solution
is gently boiled down to 10 ml and 10 ml concentrated nitric acid is added
to provide a sodium-gold nitrate. As is apparent, the nitrate is an oxidizer
and removes the chloride. The product obtained should be white crystals.
If a black or brown precipitate forms, this is an indication that there
is still Na2Au2Cl8 present. If present,
it is then necessary to restart the process at step (1).
- 20 -
(11) If white crystals are obtained, the solution is boiled to obtain just dry crystals. It is important not to overheat, i.e., bake.
(12) 5 ml concentrated nitric acid are added to the crystals and again boiled to where the solution goes to just dry. Again it is essential not to overheat or bake. Steps (11) and (12) provide a complete conversion of the product to a sodium-gold nitrate. No chlorides are present.
(13) 10 ml deionized water are added and again boiled to just dry salts. This step is repeated once. This step eliminates any excess nitric acid which may be present.
(14) Thereafter, the just dry material is diluted to 80 ml with deionized water. The solution will have a pH of approximately 1. This step causes the nitrate to dissociate to obtain NaAu in water with a small amount of HNO3 remaining .
(15) The pH is adjusted very slowly with dilute sodium hydroxide to 7.0 + 0.2. This will eliminate all free acid, leaving only NaAu in water.
(16) The NaAu hydrolyzes with the water and dissociates to form HAu. The product will be a white precipitate in water. The Au atoms have water at the surface which creates a voluminous cotton-like product.
(17) The white precipitate is decanted off from any dark gray solids and filtered through a 0.45 micron cellulose nitrate filter paper. Any dark gray solids of sodium auride should be redissolved and again processed starting at step (1).
(18) The filtered white precipitate on the filter paper is vacuum dried at 120C for two hours. The dry solid should be light grey in color which is HAuXH2O and is easily removed from the filter paper.
(19) The monoatomic gold is placed in a porcelain ignition
boat and annealed at 300C under an inert gas to remove hydrogen and to
form a very chemically and thermally stable white gold monomer.
- 21 -
(20) After cooling, the ignited white gold can be cleaned of remaining traces of sodium by digesting with dilute nitric acid for approximately one hour.
(21) The insoluble white gold is filtered on 0.45 micron paper and vacuum dried at 120C for two hours. The white powder product obtained from the filtration and drying is pure G-ORME.
The G-ORME made according to this invention will exhibit the special properties described in the "General Description" of this application, including catalytic activity, special magnetic properties, resistance to sintering at high temperatures, and resistance to aqua regia and cyanide attack.
EXAMPLE 2
RECOVERY OF METALLIC GOLD
FROM NATURALLY OCCURRING MATERIAL CONTAINING G-ORMEs
(1) 300g of dried material assayed by conventional techniques to show no gold present, ground to less than 200 mesh, is placed in a one-gallon vessel, fitted with electrodes, with 120g NaCl (Morton rock salt), 10 g KBr, and 2 liters of tap water.
(2) The anode consists of a pair of 3/8" x 12" carbon welding rods wrapped together with No. 10 copper wire. The cathode consists of 1-5/8" ID x 14" glass tube with a medium porosity glass frit (ASTM 10-15 M) with a 1" x 15" x 1/16" stainless steel strip inside in a solution of 36 g/l NaCl (approximately 500 ml). Both electrodes are placed into the sample vessel and supported by clamps extending about 5" into the sample solution.
(3) The sample is placed on a roller table at approximately
10 revolutions per minute. The electrodes are connected to a power supply
consisting of a 120 volt variac in conjunction with a 2-3 amp 400-600 PIV
rectifier. A 100 watt light bulb and the electrodes are hooked in series.
The rectifier load is connected to the anode since the rectifier
- 22 -
filters out all negative voltage and only passes positive voltage.
(4) The sample is kept under load for a period of 6-1/2 hours. The final pH is in the range of 3 - 6.5. The voltage across the electrode is 5 volts.
(5) After disconnecting the load, the sample was allowed to settle and the solution over the settled out material was removed by decantation using a peristallic pump.
(6) 800 ml of the sample was placed in a 1000 ml beaker and 20 ml concentrated sulfuric acid was added to the solution.
(7) With stirring, the solution was boiled down slowly on a hotplate until the solution was just dry. "Just dry" is as defined in Example 1. The just dry salt contains sodium gold chloride.
(8) The just dry salt was taken up in 400 ml deionized water and again boiled down to the just dry condition. There should be no discoloration at this point, i.e., a clear solution is formed.
(9) The just dry salt was then taken up in 400 ml 6M HCl, and thereafter boiled down to the just dry condition. The dilution and boiling down step was repeated four times, alternating with a deionized water and a 6M HCl wash, with the sequence controlled so that the last washing was with 6M HCl. The purpose of steps (8) and (9) is to remove all traces of hypochlorite oxidant.
(10) The just dry salts are taken up in 400 ml anhydrous ethanol and stirred for approximately ten minutes. This step is to dissolve the gold chloride salt, to remove the sodium chloride.
(11) After stirring, the slurry was filtered through #42 paper on a Buchner funnel.
(12) 5 ml of concentrated sulfuric acid was slowly added
to the filtrate, mixed, and the filtrate was then allowed to sit for approximately
one hour. The filtrate was
- 23 -
filtered through #42 filter paper on a Buchner funnel, and then passed through a filter of 0.5 micron Teflon. The sulfuric acid precipitates out any calcium. Filtration removes the precipitant and a light yellow filtrate is recovered, with all traces of calcium sulphate removed.
(13) The light yellow solution was again boiled down to just dry, taking care to avoid any charring. At this point there should be no further evaporation of ethanol and the just dry residue should be free of color. The residue should have a sweet smell similar to burnt sugar. The occurrence of the sweet smell indicates the end point of the boil-down.
(14) The just dry residue is taken up in 600 ml deionized water to provide a water-soluble gold form which is the gold auride. If desired, the G-ORME can be recovered at this stage or converted into metallic gold. For gold recovery, the solution is put into a 1000 ml beaker and an electrolysis unit was set up as shown in FIGURE 2 of the drawing.
As shown in FIGURE 2 of the
drawing, the electrolysis unit comprises a 220 volt, 120 amp power supply
(20) which is connected to the anode (12) and cathode (14) of the electrolytic
cell. The solution is stirred using a magnetic stirrer (16). The anode
(12) is a gold electrode, 2 cm2 in size, upon which gold in
solution will plate out. The cathode (14) comprises a 6.8 cm2
platinum electrode contained in a Nafion 117 chamber (18). Nafion 117 is
a perfluorocarbon sulfonic acid membrane, marketed by the duPont Company,
and is a proton-conducting membrane. Inside the Nafion chamber is 200 ml
of electrolyte solution containing 5ml sulfuric acid per 600 ml of electrolyte
solution. It is important to keep the Nafion chamber wet at all times.
The potential was measured across the electrodes and then an additional
-2.2 volts potential was applied and maintained for a period of two hours.
- 24 -
(15) After the two hours, the potential was raised to 3.0 volts and maintained for approximately 18 hours. Bubbles formed on both the gold and platinum electrodes. A black material formed on the gold electrode after three to four hours.
(16) The gold electrode was removed from solution while voltage was still being applied. The electrode was dried in a vacuum oven overnight at 115C. The electrode was weighed before and after the plating to determine the amount of gold collected.
The metallic gold is, therefore, produced from a naturally
occurring ore which, when subjected to conventional assaying, does not
test positive for gold.
EXAMPLE 3
The Preparation of Platinum Group Elements
In Monoatomic State (ORMEs) From Pure Metals
The non-metallic, monoatomic transition elements of the platinum group are prepared as follows:
(1) A selected sample of pure metal or metal salts from the group platinum, palladium, ruthenium, osmium, rhodium, or iridium are pulverized to a finely divided powder.
(2) 5.0 g of a single select elemental metal powder is intimately blended with 30 g sodium peroxide and 10 g sodium hydroxide (silica free) in an agate mortar and pestle.
(3) The blended sample is placed in a zirconium crucible and fused over a Meeker burner at maximum heat for 30 minutes.
(4) After cooling the melt, the crucible is placed into a 600 ml beaker containing 300 ml of 6M HCl.
(5) The melt should completely dissolve into the HCl.
The crucible is removed from the solution and rinsed with
- 25 -
water, and the HCl solution is carefully inspected for any insoluble metals or metal oxides which, if present, must be filtered out and fused again as in step (2) above.
(6) The HCl solution is gently boiled down to just dry salts. "Just dry" is as defined in Example 1.
(7) The just dry salts are taken up in 300 ml of pH 1 HCl solution and then gently boiled down to salts again. The salts at this point, depending on the selected metal sample, are alkali chlorides together with alkali-cluster-noble metals-metal chlorides.
(8) The procedure of steps (6) and (7) is repeated four times, being careful not to bake the salts.
(9) The salts are diluted with 400 ml of deionized water.
(10) 30 ml of concentrated perchloric acid is added to the solution and then slowly boiled to fumes of perchloric acid.
(11) Steps (9) and (10) are repeated three additional times. If the solution salts out before fuming is achieved, it is necessary to add an additional 5 ml of perchloric acid to replace acid lost in fuming. If ruthenium or osmium is the select metal, steps (10), (11) and (12) must be carried out under reflux and washed back with water since ruthenium and osmium will volatilize. The salts at this point, depending on the selected metal sample, are alkali monoatomic noble metal oxides.
(12) The salts are diluted to 400ml with deionized water.
(13) The pH is adjusted very slowly with sodium hydroxide solution until the solution maintains the pH of 7.0 + 0.2 for more than 12 hours.
(14) Boil the solution for several hours, adding deionized
water to maintain 400 ml during the entire boiling until a reddish-brown
hydroxide precipitant is formed which is filtered on a fine fritted, glass
filter.
- 26 -
(15) The hydroxide precipitant is dissolved off the fritted glass filter with 400ml of pH 1 HCl and then boiled for approximately ten minutes. If the sample contains rhodium or iridium, sodium bromate should be added as an oxidant prior to boiling.
(16) The solution is neutralized slowly with sodium bicarbonate to pH 7, and the solution is boiled again and allowed to cool.
(17) The precipitant which is formed is filtered again through a fine fritted glass filter. The material at this point, depending on the selected metal sample, is a monoatomic noble element hydroxide.
(18) The hydroxide together with the filter are vacuum dried at 120C for approximately 12 hours.
(19) The dried material is carefully transferred from the filter to a quartz ignition boat.
(20) The ignition boat is placed in a cold tube furnace and the temperature is slowly (2C/min) raised under a hydrogen atmosphere to 600C and held at this temperature for one hour and then slowly (2.5C/min) cooled down to room temperature under hydrogen and then the sample is purged with argon for approximately one hour to remove occluded hydrogen. The material, an ORME, will be a greyish-black powder and will be completely amorphous to x-ray analysis. In other words, a certified pure noble metal powder has been converted to a "non-analyzable" form.
At this point the ORMEs, depending upon the selected element sample, will be orbitally rearranged due to the d orbital hole or holes, i.e., positive hole(s). The ORMEs are identified as having an infrared doublet between 1400 and 1600 cm-1. The doublet indicates the presence of an electron pair moving between the d and s orbitals.
These materials have a number of applications as previously
described, one of which is as catalysts in an electrochemical cell.
- 27 -
EXAMPLE 4
PROCEDURE FOR SEPARATION OF PLATINUM GROUP
ELEMENTS (PGEs) FROM ORE CONTAINING ORMEs
The class of ores which are processed to form ORMEs, when analyzed by conventional instruments normally used for determination of Platinum Group Metals (PGM), will indicate that essentially no metals of this PGM group are present.
In the separation of the PGE from the ore, the pretreatment of the ore sample is crucial. If the sample is not prepared properly, the PGEs in their ORME state are virtually impossible to separate. The separated elements are not necessarily in an ORME state.
The purpose of the pretreatment is primarily for the removal of silica. Pretreatment comprises crushing and pulverizing the ore to a fine powder (-200 mesh). A sample of 50 g of the pulverized ore and 100 g ammonium bifluoride, NH4HF2, are weighed and placed in a 1000 ml Teflon beaker. The ore and NH4HF2 are moistened with distilled water and approximately 200 ml HF (hydrofluoric acid) is added. The sample is baked to dryness on a hotplate. This procedure is repeated four times each with more HF. The sample is transferred to a platinum dish and roasted over a hot flame until the sample turns a dull red-brown color. After this treatment, most of the silica has been removed as H2SiF6 (white fumes that evolve during roasting).
The sample is now placed in a zirconium crucible with
200 g NaNO3 (sodium nitrate) and 500g Na2CO3
(sodium
carbonate). The sample is then fused using a Fisher burner and a propane
torch to a red hot melt. When cool, the fusion should be an aquamarine
color, or a light brown color. The light brown color means the sample has
passed through the aquamarine stage. This poses no problems in the subsequent
separation and determination of the PGEs. If the melt cools to a light
green color, fusion is not com-
- 28 -
plete. It must be fused again until it reaches the aquamarine end point.
In the zirconium crucible containing the cooled melt, place an "X" shaped Teflon coated stirring bar and minimum amount of distilled water. Place the crucible in a beaker and cover with a watch glass. Place the beaker on a stir plate to slurry/dissolve the sample from the crucible. A minimum amount of distilled water should be used in the removal. The sample is now ready for distillation.
(1) DISTILLATION AND SEPARATION OF OSMIUM AND RUTHENIUM
The first PGEs are separated by a perchloric acid distillation with ruthenium and osmium being distilled off as RuO4 and OsO4. Platinum, palladium, rhodium, and iridium are left in the pot liquor. The distillation apparatus in diagrammatic form is illustrated in FIGURE 3 of the drawing, as used on a 5 g sample of ore.
Referring to FIGURE 3 of the drawing,
Flask #1 has a 500 ml volume and contains 5g of ore in 250 ml of solution/slurry.
Flask #2 has a 250 ml volume with 60 ml 1:1 HCl and 15 ml 30% H2O2.
Flask #3 has a 50 ml volume with 20 ml 1:1 HCl and 15 ml 30% H2O2.
Flask #4 has a 200 ml volume with 100 ml 1:1 HCl saturated with SO2 (sulfur dioxide).
Flasks #5 and #6 have a 100 ml volume with 60 ml 1:1 HCl saturated with SO2.
The flasks are all interconnected with glass conduits and ground glass ball and socket joints.
The distillation proceeds as follows: A closed system
is used with N2 (nitrogen) as a carrier gas for RuO4
and OsO4. To Flask #1 60 ml of 70 % HClO4 (perchloric
acid) is added slowly from the separatory funnel 10. Once all of the HClO4
is added, the flask is heated. At a temperature
- 29 -
of 105-112C, a white cloud is seen flowing into Flask #2. The heat is continued until fumes of HClO4 begin to come off at approximately 175C. The heating is continued to 210C when the temperature stops rising. The system is then cooled to 100C. At this point 20 ml of 70% HClO4 and 20 ml distilled water are added to Flask #1, again through the separatory funnel; and the system is heated to 210C again, then cooled again to 100C. 10 ml of 70% HClO4 and 10 ml distilled water are added to Flask #1 and the sample is heated again to 210C. The distillation is repeated once more as before.
After the fourth distillation, the heat on Flask #1 is turned off and heat is applied to Flask #2, bringing it to a boil slowly to drive any OsO4 out of the RuO4 fraction. Nitrogen purge gas is still flowing and must be controlled to prevent back flow. Boiling is continued until Flask #3 is almost full or the H2O2 has been almost driven out of Flask #3. The presence of H2O2 is indicated by tiny bubbles forming all over the glass surface. The entire system is then cooled to room temperature, with the nitrogen gas flowing continuously through the cool down.
The distillation receiving flasks are then dismantled.
Flask #4, #5, and #6 contain the osmium fraction as OsO4. These
are combined in a 600 ml beaker. Flask #2 and #3 contain the ruthenium
fraction as RuO4 and are combined in a 600 ml beaker. The contents
of Flask #1 which contains platinum, palladium, rhodium, and iridium are
retained in the distillation flask to remove HClO4 by heating
to dryness as described in Section 4. These fractions are now ready for
further analysis and separation. The osmium and ruthenium fractions must
sit in solution at room temperature for 16-24 hours before continuing with
the steps (2) and (3).
- 30 -
(2) SEPARATION OF OSMIUM
The osmium distillate after sitting for 16-24 hours at room temperature is processed as follows: The osmium fraction from the distillation is slowly evaporated to approximately 10 ml of solution. Then 25 ml of concentrated HCl (hydrochloric acid) are added and the sample is again evaporated to approximately 10 ml. This is repeated five times. On the last digestion, the sample is carefully taken to moist salts at which point it is diluted to 200 ml with distilled water and brought to a boil. The hot solution is filtered through #42 Whatman paper, washing with a minimum amount of 0.1 N HCl.
After cooling to approximately 40C, the pH of the sample is then slowly adjusted on a calibrated pH meter using a saturated solution of NaHCO3 (sodium bicarbonate), to a pH of 4 while stirring vigorously. The solution then is gently boiled for 5-10 minutes, removed from the heat, and let stand for a period of at least twelve hours. The osmium precipitates are a reddish-brown hydrated dioxide.
The solution is filtered through a dry, tared porcelain filter crucible using the Walters crucible holder. Most of the solution is decanted through the filter crucible, being careful not to disturb or float the precipitate. The filter should not pull dry. Pour the last 100-200 ml of solution containing precipitate in the filter. Be prepared to immediately rinse the precipitate with hot 1% w/v NH4Cl solution (filtered through 0.45 micron pad during preparation). A wetted rubber policeman is used to thoroughly scrub the beaker and rinse after each scrub with hot 1% NH4Cl.
The crucible is dried overnight at 105C in a vacuum oven. The cooled, dry crucible is weighed and the approximate osmium value is calculated from this OsO2 weight.
With the crucible on vacuum again, the precip-
- 31 -
itate is rinsed with two aliquots of 20 ml each saturated NH4Cl solution. Leave 100-200 mg of the solid NH4Cl on the precipitate. Dry gently in a vacuum oven for 1-2 hours at 100C.
The sample is now ready for tube furnace hydrogen reduction. Place the filter crucible on its side in a quartz tube, and insert the tube into the furnace center. Start argon and hydrogen gas flow through the furnace. Allow the temperature to increase slowly to dehydrate the precipitate without igniting it. Decrease the argon flow until only hydrogen flows. Then heat at 360-375C until all NH4Cl is sublimed.
Continue heating the precipitate in hydrogen only at 500C for 20 minutes to complete reduction to osmium metal. Cool the crucible in hydrogen to ambient temperature. Replace hydrogen with carbon dioxide for 20 minutes to prevent any oxidation when the reduced metal is first exposed to air. Weigh as elemental osmium.
(3) SEPARATION OF RUTHENIUM
The ruthenium distillate after sitting 16-24 hours at room temperature is processed as follows: The ruthenium fraction from the distillation is slowly evaporated to approximately 10 ml of solution. Then 25 ml of concentrated HCl are added and the sample is digested again to approximately 10 ml. This procedure is repeated five times. On the last digestion, the sample is carefully taken to moist salts on a steam bath. The sample must not be hot enough for HClO4 traces to reoxidize the ruthenium. Add 200 ml of distilled water, and bring the solution to a boil. Filter the hot solution through No. 42 Whatman paper, washing with a minimum amount of 0.1 N HCl.
After cooling to approximately 40C, the pH of the sample
is slowly adjusted on a calibrated pH meter with a saturated solution of
NaHCO3 to pH 6 while stirring vigorously. The solution is brought
to a gentle boil for 5-10
- 32 -
minutes before removing it from the heat. The sample is permitted to stand for a period of at least twelve hours. The ruthenium precipitates as a yellowish-brown hydrated dioxide.
The solution is filtered through a #42 Whatman ashless filter paper wetted with 1% w/v (NH4)2SO4 (filtered through a 0.45 micron pad during preparation). Decant most of the solution through the filter paper, being careful not to disturb or float the precipitate. Pour the last 100-200 ml of solution containing most of the hydrated oxide in the paper all at once. A wetted rubber policeman is used to thoroughly scrub the beaker. A piece of #42 ashless filter paper wetted with 1% w/v (NH4) 2SO4 is used to complete the transfer. The precipitate is washed twice with hot 1% w/v (NH4)2SO4 and once with hot 2.5% w/v (NH4)2SO4. The filter is allowed to drain as dry as possible.
The paper is transferred to a tared quartz boat, and dried gently in an oven at 110C.
The boat is placed in a quartz tube for final firing and reduction in the tube furnace. From a cold start (below 100C), pass enough air over the sample to ignite the paper without mechanical loss of precipitate. Increase the furnace temperature slowly to 500C and maintain this temperature until the paper ignition is complete. Pull the boat out of the heated section and allow it to cool to 150C or less. Purge the tube with argon, then hydrogen. Complete the hydrogen reduction with sample in the heated section at 500C, then to 600C for 20-30 minutes.
Pull the sample out of the heated section to cool to less than 100C with hydrogen being passed over the sample. Complete the cooling with carbon dioxide to ambient temperature (approximately 10-15 minutes).
The cooled ruthenium is washed twice with 1% w/v (NH4)2SO4
to dissolve the last traces of soluble salts. Ignite again in air and hydrogen
as described above. Weigh as elemental ruthenium.
- 33 -
(4) SEPARATION OF PLATINUM
The platinum, palladium, rhodium, and iridium fraction in HClO4 from the distillation is evaporated to dryness in a beaker. The procedure takes considerable time and care since HClO4 is being fumed off. When the sample reaches a dry salt state and is cooled, distilled water and concentrated HCl are added, and the sample is evaporated again. The water, HCl treatment is repeated twice more. After the sample has been evaporated for the last time, it is diluted with distilled water to 300 ml. The sample is now ready to separate platinum from rhodium, palladium, and iridium. At this stage either an ion-exchange process, which is designed for production of larger quantities of separated ORMEs, or a non-precise quantitative separation may be used. The following procedure details the quantitative separation.
The sample is brought to a boil and 200 ml of 10% w/v NaBrO3 (sodium bromate) solution are added and the sample is boiled again. When the sample has reached boiling, it is removed from the heat, cooled to 40C, and the pH is adjusted with a calibrated pH meter to pH 6 with a saturated NaCHO3 solution. 100 ml of 10% NaBrO3 are added and the solution is brought to a gentle boil for 15 minutes. The sample is then cooled and the precipitate is allowed to coagulate for 20-30 minutes.
The sample is then filtered on a medium porosity fritted
glass filter and washed with 1% NaCl solution pH 6.5 - 7.5 (filtered during
preparation through a 0.45 micron pad). The filtrate contains the platinum
and the precipitate contains palladium, rhodium, and iridium as PdO2,
RhO2 and IrO2 in hydrated form. The precipitate is
redissolved with 6N HCl, boiled and reprecipitated as above two or more
times to ensure complete separation of platinum from palladium, rhodium,
and iridium.
- 34 -
The filtrates from the three precipitations are combined in a 1000 ml beaker and 50 ml of concentrated HCl are added. The sample is boiled to dryness to remove bromine and any traces of HClO4 that still might be present. Add 50 ml of water and 50 ml concentrated HCl. Boil to dryness again and repeat two more times, with the last time being to provide moist crystals rather than boiling to dryness. The sample is diluted to 200 ml with distilled water and 40 ml of HCl are added.
The sample is heated to a gentle boil and a stream of H2 (hydrogen) gas is passed through the sample for ten minutes, followed by passing a stream of H2S (hydrogen sulfide) gas through the solution while continuing with a flow of H2. The solution is allowed to cool while H2S is passing though it. The platinum precipitates as brown black PtS2.
The solids are filtered through #42 Whatman ashless filter paper and the precipitate washed with 1% v/v HCl. The filter and precipitate are transferred to a tared porcelain crucible. The filter is dried gently, then the residue ignited in air to red heat using a Meeker burner. The metal residue is leached with 1% v/v HCl and washed onto a second #42 ashless filter paper. The residue is washed thoroughly with hot distilled water. The filter is transferred to the same porcelain crucible, dried, and heated to red heat using a Meeker burner. The residue is weighed as platinum metal. The PtS2 precipitate can also be reduced under H2 in the tube furnace.
(5) SEPARATION OF PALLADIUM
The precipitate of hydrated dioxides of palladium, rhodium,
and iridium remaining from step (4) are dissolved in 1000 ml of 6 N HCl
and diluted to 4000 ml with distilled water. The sample is then filtered
on a 0.45 micron filter. To the solution is added a sufficient volume of
1% w/v dimethylglyoxime in 95% ethanol (250 ml) to precipitate all the
palladium with gentle boiling. The sample is set aside
- 35 -
for a minimum of one hour, then filtered into a tared porcelain filter crucible. Wash with 0.1 N HCl and then with water. The filtrate is retained for rhodium and iridium separation. The precipitate is dried at 1100C and the yellow precipitate is weighed as palladium dimethylglyoxime, with palladium being 31.67% w/w of the total precipitate.
(6) SEPARATION OF RHODIUM
The filtrate from the first palladium precipitation is diluted to 500 ml and 10 ml of concentrated H2SO4 and 10 ml of concentrated HNO3 are added. The filtrate is evaporated with heat until heavy fumes of H2SO4 are evolved. After cooling, 10 ml concentrated HNO3 are added and again heated until fumes are evolved. This treatment is repeated until no more charring results and all organic material has been destroyed. The solution remaining is cooled and 20 ml water are added. Evaporation with heating to heavy fumes is again repeated. The water wash is repeated two times to destroy any nitroso compounds that might interfere in the rhodium determination.
The solution is diluted to 200 ml and heated to boiling. A solution of 20% TiCl3 (titanous chloride) is added dropwise until the solution retains a slight pink color. Boil the solution for two minutes, cool, and filter the solution through Whatman #42 ashless filter paper. If any rhodium has precipitated out, wash the paper with 0.9 N H2SO4. Then char the filter paper in a 5 ml concentrated H2SO4. Add 5 ml HNO3 to heat and destroy organic matter as previously described. Dilute the solution with 50 ml water and combine with the filtrate from the TiCl3 precipitation.
The rhodium is separated from the iridium by removal of
the excess titanium in a cupferron extraction with chloroform. The solution
is chilled in an ice bath and placed in a 500 ml separatory funnel. To
this 5 ml aliquots of chilled 6% aqueous cupferron are added, giving a
milky yellow solution. If the cupferron solution is dar-
- 36 -
kened, it should be treated with activated charcoal and filtered through a 0.45 micron pad. The titanium is extracted in 25 ml aliquots of cold chloroform. The extract is a clear yellow solution which is poured into a waste container. When no more yellow color is extracted, another 5 ml aliquot of cupferron solution is added. After many aliquots to remove the yellow titanium cupferrate, the extract turns a red brown. This fraction is collected in a separate beaker as the rhodium fraction. All extractions following this are added to the rhodium fraction in a 600 ml beaker. The extraction is complete when an aliquot of cupferron turns the solution milky white and the chloroform extract is clear to very light green. Retain the solution for iridium separation.
The extract is evaporated to dryness separating the chloroform from the rhodium fraction. 50 ml of aqua regia are added and the sample is evaporated to dryness to destroy organic material. Add 10 ml concentrated H2SO4 and 10 ml HNO3 and heat to fumes. Repeat HNO3 treatment until no more charring results and all organic material has been destroyed. The solution is cooled and 20 ml water is added, followed by evaporation to heavy fumes again. Repeat the water wash two times to destroy any nitroso compounds.
The sample solution is diluted to 200 ml with water. Then 10 ml of 10% NaBrO3 is added and the sample is heated to boiling. The sample is then cooled to 40C and the pH adjusted to pH 6.0 with NaHCO3. 10 ml of NaBrO3 are added and the sample heated to a boil. The sample is cooled and filtered on a weighed porcelain crucible. The sample is dried in a vacuum oven and the precipitate is weighed as RhO2.
The material is then purified by dissolving the RhO2 precipitate from the weighing crucible with 6 N HCl and evaporate to moist salts and proceed as above.
The rhodium oxide is removed from the weighing crucible
by using a 20% v/v H2SO4 solution. Then dilute the
- 37 -
solution to 200 ml with water, and heat to boiling. Add dropwise a solution of 20% TiCl3 until the solution retains a slight pink color while boiling. A precipitate of rhodium will form. Allow the solution to cool to 40C. If it loses color, boil and add more TiCl3. If color remains, filter through Whatman #42 ashless filter paper. The precipitate is washed with hot 10% v/v H2SO4 until the filtrate ceases to show the orange titanium complex with H2O2, then wash twice more.
Redissolve the rhodium as before to destroy the organic material. Add 10 ml concentrated H2SO4 and 10 ml of HNO3 to char the paper. Repeat the HNO3 treatment until no more charring results and all organic material has been destroyed. Cool the solution, add 200 ml water, and evaporate to heavy fumes again. Repeat the water treatment two times to destroy any nitroso compounds.
Add 20 ml of water and 10 ml of concentrated HCl. Gently boil the solution 15 minutes to get the rhodium into the state from which it can be precipitated as a sulfide. During treatment the color of the solution will change from yellow to rose. Filter the solution #42 Whatman filter paper and wash with 1% v/v HCl. Dilute the solution to 400 ml with water.
Precipitate the rhodium as sulfide from the solution kept at the boiling point by passing a rapid stream of H2S (hydrogen sulfide) gas through it. Allow the solution to cool with H2S passing through it. Allow the brown-black rhodium sulfide to settle.
Filter the produce sulfide through #42 Whatman ashless filter paper. Wash with 2.5% v/v H2SO4 and finally with 1% v/v HCl. Finally, dry the filter paper gently in a tared quartz boat.
Place the boat in the quartz tube for final firing and
reduction in the tube furnace. From a cold start (below 100C), pass enough
air over the sample to ignite the paper
- 38 -
without mechanical loss of precipitate. Increase the furnace temperature slowly to 500C and maintain this temperature until paper ignition is complete. Then complete the air firing at 900C for 20 minutes. Pull the crucible out of the heated section and allow it to cool to 200C or less. Purge the tube with argon, then hydrogen. Complete the hydrogen reduction with sample in the heated section at 900C for 20-30 minutes.
Pull the sample out of the heated section to cool to less than 100C, with hydrogen being passed over the sample. Complete the cooling with carbon dioxide to ambient temperature for 10-15 minutes.
Wash the cooled rhodium twice by decantation with cool 1% w/v (NH4)2SO2 to dissolve the last traces of soluble salts. Dry gently, ignite again in air and hydrogen as described above. Weigh as elemental rhodium.
(7) SEPARATION OF IRIDIUM
The solution left in the separatory funnel from the cupferron extraction contains the iridium. Transfer it quantitatively with a 1% v/v H2SO4 wash to a 600 ml beaker. Add 10 ml of concentrated HNO3. Evaporate to heavy fumes of H2SO4. Cool, add 10 ml more HNO3 and again heat to fumes. Repeat this treatment until no more charring results and all organic material has been destroyed. Cool the solution, add 20 ml water and evaporate to heavy fumes again. Repeat with the water treatment two times to destroy any nitroso compounds. Dilute with water to 300 ml.
Bring the sample to a boil and add 20 ml of 10% w/v NaBrO3
solution and boil again. When the sample has reached boiling, it is removed
from the heat, cooled to 40C, and the pH is adjusted with a calibrated
pH meter to 7 with saturated NaHCO3 solution. Add 10 ml of 10%
NaBrO3 and bring to a gentle boil for 15 minutes. The sample
is then cooled slowly and the precipitate is allowed to coagulate for 20-30
minutes.
- 39 -
The precipitate is filtered into a tared porcelain crucible in a Walters crucible holder. Decant most of the solution through the filter crucible, being careful not to disturb or float the precipitate. Do not let the filter pull dry. Pour the last 10-20 ml of solution containing the precipitate into the filter. Be prepared to immediately rinse and police and beaker with 10% w/v NaC1 solution. Dry the filter at 110C under vacuum for 1-2 hours. Dissolve the precipitate with 6N HCl and evaporate to moist salts and proceed as before, for a cleaner iridium fraction.
Wet the precipitate with saturated NH4Cl solution and approximately 100 mg of solid NH4Cl. Dry gently in a vacuum oven again at 110C for 1-2 hours.
The sample at this point, which is the hydrated iridium ORME can be treated by alternate procedures. In the first procedure the sample will be treated to provide an iridium S-ORME and then utilized to establish the existence of a Meissner field, a property unique to superconducting materials. In the second procedure, the sample will be treated so as to form elemental iridium.
Procedure A
The iridium fraction is placed in a quartz ignition boat
and the boat inserted into a tube furnace for slow reduction under hydrogen
gas. The hydrogen gas is flowed slowly over the sample maintaining a slight
positive pressure in the tube at all times. The temperature of the tube
furnace is raised very slowly and uniformly up to 850C, taking care not
to allow the heating rate to exceed 2C per minute. The 850C temperature
is maintained for one hour, then the sample is slowly cooled under hydrogen
gas, being careful not to exceed a 2.5C reduction in temperature per minute
until room temperature has been achieved. Nitrogen gas is then introduced
into the tube and the hydrogen gas is shut off. The tube is then purged
for eight hours with nitrogen gas. The sample at this point will be a grey-black
- 40 -
amorphous powder. The powder is removed from the tube and then placed in a protected area so that it can react with air for at least two days (48 hours).
Approximately 10 mg of the resultant powder is transferred to a controlled atmosphere bifilar-wound heating element Thermo Gravimetric Analysis (TGA) instrument (Perkin-Elmer Thermal Analysis (PE/TGS-2), Temperature Programmer (PE/System 4), Thermal Data Station (PE/TADS). And Graphics Plotter (PE/THERM PLTTR). The sample is heated in the instrument at the rate of 1.2C per minute under an atmosphere of helium gas to 850C, and then immediately cooled at 2C per minute to room temperature. The heating and cooling cycles are repeated four times.
The bifilar winding of the heating element possesses an extremely small magnetic field in that the weighed sample can never be exactly equal distance from both wires due to the winding configuration. The depolarized field will not react with ordinary metal samples or normal magnetic (N-S polarized) materials. However, a superconductor will react with an external magnetic field, even one of small magnitude.
FIGURES 8-17, which are weight/temperature
plots of alternate heating and cooling of the iridium S-ORME sample material
over five cycles, depict the Meissner field generation and the frequent
collapsing and regeneration of the field. Specifically, FIGURE
8, Plot IR1H1, demonstrates the first heating cycle which establishes
approximately a 26% weight loss. This weight loss is primarily due to loss
of water. FIGURE 9, Plot IR1C1, read from the
right to the left with 100% being the 75% of Plot IR1H1 (FIGURE
8), demonstrates weight gain and flux jumping upon cooling. The apparent
weight gain and flux jumping establishes that the material is superconductive.
A material such as iron which is not superconductive would show a plot
which is essentially a flat line. The remaining plots, i.e., FIGURES
10-17, showing the effect of alternate heating and cooling, estab-
- 41 -
lish that each treatment extends the Meissner field generation in the direction of room temperature. FIGURE 17, Plot IR1C5, shows the flux jumping very close to room temperature.
The sample, after the above annealing treatment has been completed, will be white in color. The white powder is chemically inert to normal oxidation-reduction chemistries. It does not gain weight readily on exposure to air. However, gases such as nitrogen, oxygen, carbon monoxide, and carbon dioxide do apparently adsorb to the surface resulting in "flux pinning" as the term is used in describing behavior of superconducting materials of the S-ORME.
Procedure B
The sample is subjected to furnace ignition and hydrogen reduction. Place the filter crucible on its side in the quartz tube and insert into the tube furnace center. Start the air flowing gently. Allow the temperature to increase slowly to dehydrate the precipitate completely. Heat until all NH4Cl is sublimed at 360-375C. Continue heating in air to 800C.
Remove the crucible from the heated section of the furnace and cool to 200C or less. Purge the tube with argon, then hydrogen. Complete the hydrogen reduction of the sample in the heated section at 800C for 20-30 minutes.
Pull the sample out of the heated section to cool to less than 100C while hydrogen is being passed over the sample. Complete the cooling by treatment with carbon dioxide for 10-15 minutes to ambient temperature.
Wash the cooled iridium with 1% w/v (NH4)2SO4
- 42 -
twice to dissolve the last traces of soluble salts. Dry gently, ignite again in air and hydrogen as described above. Weigh as elemental iridium, or the Ir-ORME. If the sample is partially dissolved in aqua regia in preparation for an Inductively Coupled Plasma Mass Spectroscopy (ICP-MS) testing, then the instrument will indicate the presence of metallic iridium. In other words, prior to treatment of the ore, conventional assay techniques indicated that no iridium was present. After treatment and separation of the ORMEs, a slow reduction under hydrogen gas, followed by aqua regia treatment, will convert part of the Ir-ORMEs into their constituent T-metal.
As will be apparent to one skilled in the art, various
modifications can be made within the scope of the aforesaid description.
Such modifications being within the ability of one skilled in the art form
a part of the present invention and are embraced by the appended claims.
- 43 -
THE CLAIMS DEFINING THE INVENTION ARE AS FOLLOWS:
THE CLAIMS:
1. In a separated and substantially pure, stable form, a non-metallic, orbitally rearranged monoatomic transition or noble metal element selected from the group consisting of cobalt, nickel, copper, silver, gold, palladium, platinum, ruthenium, rhodium, iridium, and osmium having a d orbital hole sharing energy with an electron or electrons, said shared energy identified as a doublet in an infrared spectrum of from between about 1400 and 1600-1 cm.
2. The orbitally rearranged monoatomic element of claim 1 wherein said element is gold.
3. The orbitally rearranged monoatomic element of claim 1 wherein said element is silver.
4. The orbitally rearranged monoatomic element of claim 1 wherein said element is copper.
5. The orbitally rearranged monoatomic element of claim 1 wherein said element is palladium.
6. The orbitally rearranged monoatomic element of claim 1 wherein said element is platinum.
7. The orbitally rearranged monoatomic element of claim 1 wherein said element is ruthenium.
8. The orbitally rearranged monoatomic element of claim 1 wherein said element is rhodium.
9. The orbitally rearranged monoatomic element of claim
1 wherein said element is iridium.
- 44 -
10. The orbitally rearranged monoatomic element of claim 1 wherein said element is osmium.
11. The orbitally rearranged monoatomic element of claim 1 wherein said element is cobalt.
12. The orbitally rearranged monoatomic element of claim 1 wherein said element is nickel.
13. Process of forming a non-metallic, orbitally rearranged monoatomic form of an element selected from the group consisting of cobalt, nickel, copper, silver, gold, palladium, platinum, ruthenium, rhodium, iridium, and osmium from the corresponding element in metal form comprising treating said metal form by forming a salt thereof, exhaustively solubilizing and evaporating said salt in an aqueous medium until a diatom of said metal form is obtained; and thereafter treating said diatom with an alkali metal in the presence of water to form said orbitally rearranged, stable monoatomic form of said element.
14. Process of forming a metal selected from the group
consisting of cobalt, nickel, copper, silver, gold, palladium, platinum,
ruthenium, rhodium, iridium, and osmium from a material having the corresponding
element present in a non-metallic, orbitally rearranged monoatomic stable
form of said element, comprising separating said element in said orbitally
rearranged monoatomic form from said material, and then subjecting said
separated, non-metallic, orbitally rearranged mono-atomic stable form to
a two-step negative potential of at least 1.8 to 2.2 V initially, and then
to at least 2.5 V until the said element is formed by electroplating techniques.
- 45 -
15. Process of forming a metal selected from the group consisting of cobalt, nickel, silver, palladium, platinum, ruthenium, rhodium, iridium, and osmium from a material having the corresponding element present in a non-metallic, orbitally rearranged monoatomic stable form of said element, comprising subjecting said element in said orbitally rearranged monoatomic stable form to a treatment with nitric oxide at elevated temperatures.
16. Process of treating the stable non-metallic, orbitally rearranged monoatomic transition or noble metal element of claim 1 by subjecting said element to alternate heating and cooling cycles under an inert gas and supplying an external magnetic field to said element until said element no longer exhibits a doublet in the infrared spectrum and exhibits magnetic flux exclusion at temperatures above 200K.
17. The product formed by the process of claim 16.
18. An orbitally rearranged monoatomic element, selected from cobalt, nickel, silver, palladium, platinum, ruthenium, rhodium, iridium, and osmium having a d orbital hole sharing energy with an electron or electrons: having a doublet in its infrared spectrum between 1400 and 1600 cm-1; having non-metallic characteristic, and being in substantially pure form.
19. An orbitally rearranged monoatomic element in substantially
pure form and substantially as herinbefore described.
- 46 -
20. A process of forming a non-metallic, orbitally rearranged monatomic form of an element selected from cobalt, nickel, silver, palladium, platinum, ruthenium, rhodium, iridium, and osmium substantially as herein described in any one of the Examples.
21. A non-metallic, orbitally rearranged monoatomic form of an elements selected from cobalt, nickel, silver, palladium, platinum, ruthenium, rhodium, iridium, and osmium prepared by the process claimed in claim 13 or 20,
22. A process of forming a metal from a non-metallic, orbitally rearranged monoatomic form of an element selected from cobalt, nickel, silver, palladium, platinum, ruthenium, rhodium, iridium, and osmium substantially as hereinbefore described.
23. A metal formed by the process claimed in claim 14,
15 or 22.
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP program to upload the entire file without some padding at the end like this. If you have any ideas about how to solve this problem contact me at sumro@zz.com
This is padding because I cannot get this FTP
program to upload the entire file without some padding at the end like
this. If you have any ideas about how to solve this problem contact me
at sumro@zz.com
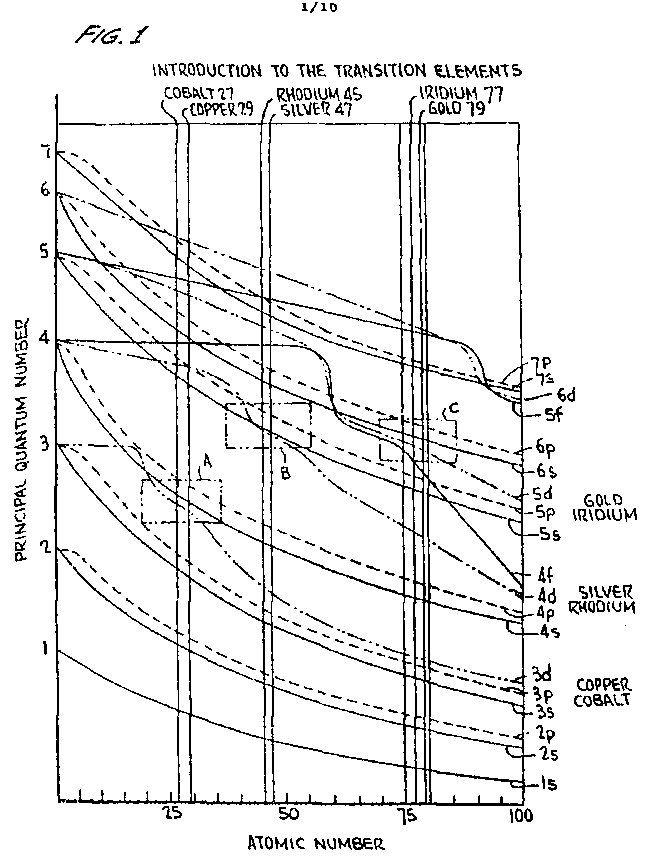{kind=link}
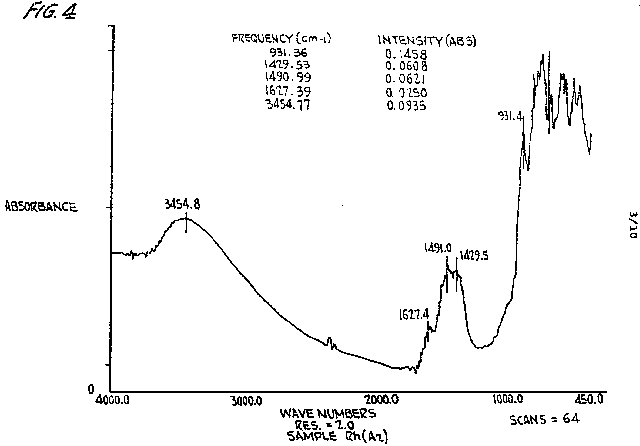{kind=link}
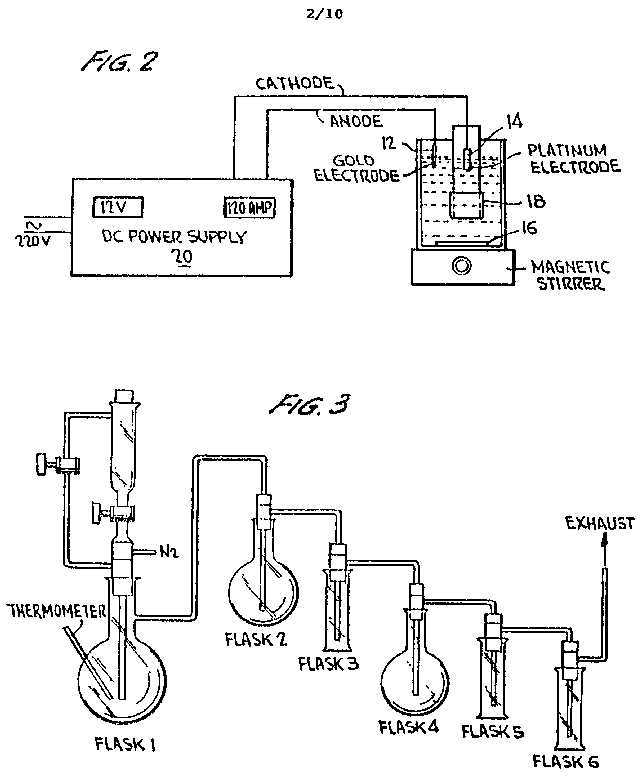{kind=link}
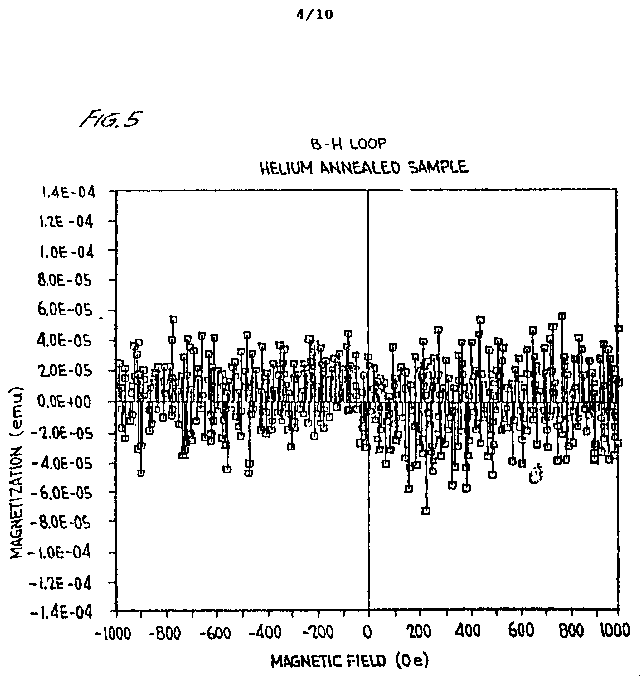{kind=link}
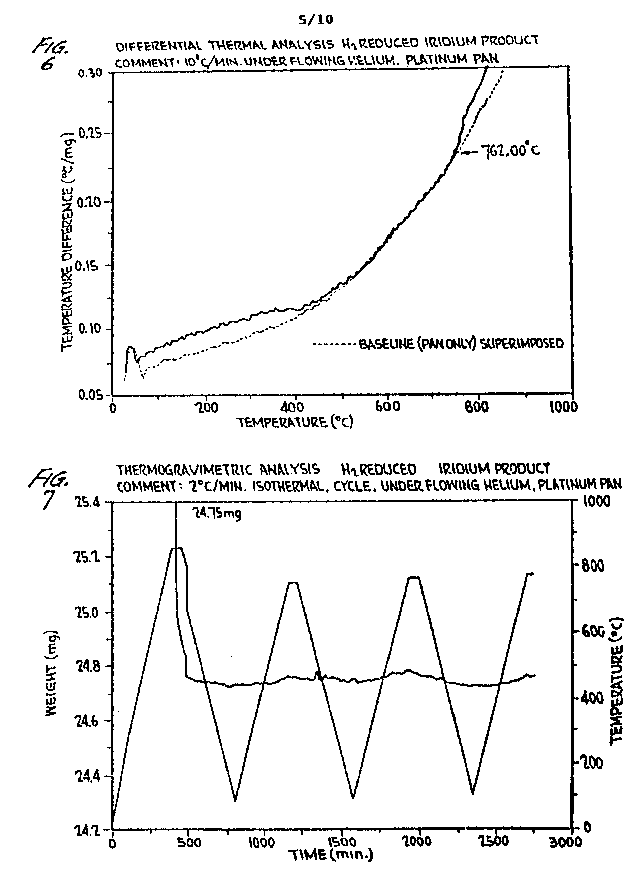{kind=link}
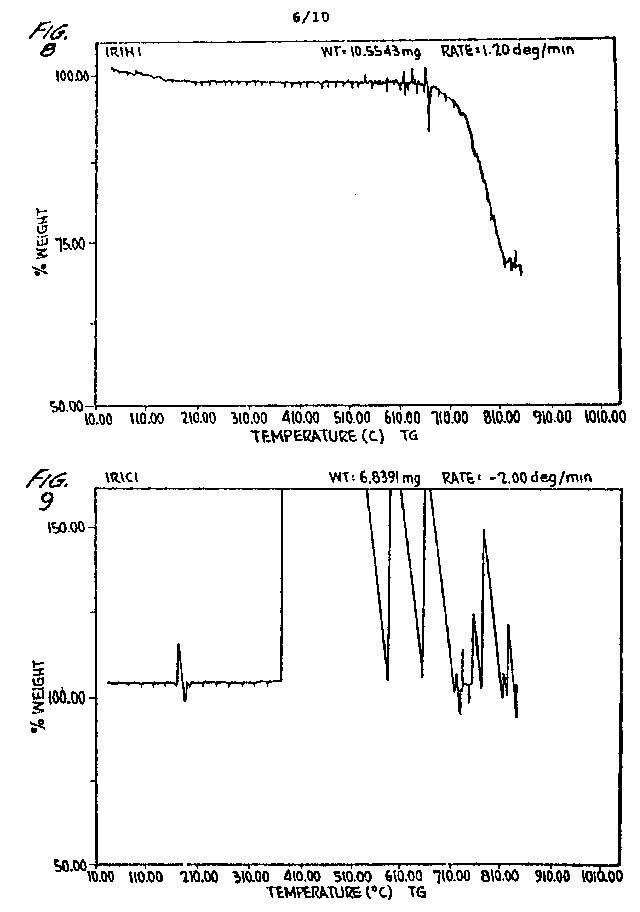{kind=link}
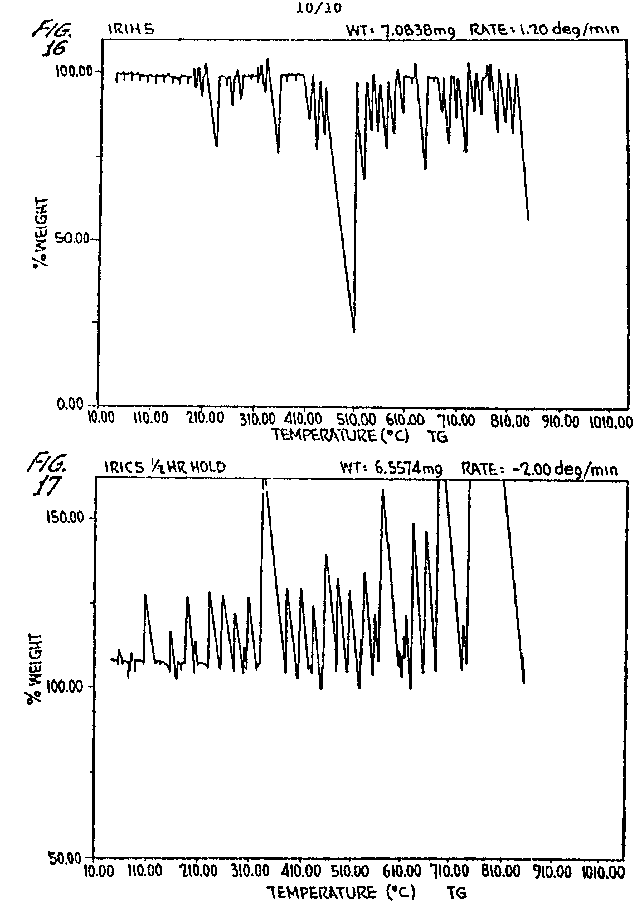{kind=link}